Protonation reactions, i.e., A + H 
 AH , are
among the most important in chemistry and biology.
Protonation/deprotonation is the first
step in many fundamental chemical rearrangements and in most
enzymatic reactions.
Two quantities are used to characterize the ability of a molecule
in the gas phase to accept a proton. The gas phase basicity is the
negative of the free energy change associated with the reaction.
The more frequently used index,
the proton affinity, is the negative of the enthalpy change at standard
conditions. Experimental
determination of these parameters is not easy (for an excellent review
on this topic see Dixon and Lias
AH , are
among the most important in chemistry and biology.
Protonation/deprotonation is the first
step in many fundamental chemical rearrangements and in most
enzymatic reactions.
Two quantities are used to characterize the ability of a molecule
in the gas phase to accept a proton. The gas phase basicity is the
negative of the free energy change associated with the reaction.
The more frequently used index,
the proton affinity, is the negative of the enthalpy change at standard
conditions. Experimental
determination of these parameters is not easy (for an excellent review
on this topic see Dixon and Lias  ), and with the phenomenal growth in
computer power in recent years,
much attention has been given to the possibility of
calculating these parameters by quantum methods. Ab initio
approaches are very successful in providing reliable values of proton
affinities and gas phase basicities for small molecules even at lower
levels of theory
), and with the phenomenal growth in
computer power in recent years,
much attention has been given to the possibility of
calculating these parameters by quantum methods. Ab initio
approaches are very successful in providing reliable values of proton
affinities and gas phase basicities for small molecules even at lower
levels of theory  . However, due to computational
expense, application of ab initio methods to the estimation of
proton affinities is still impractical for larger molecules.
Semiempirical methods such as AM1, MNDO and PM3, are not consistently
reliable
in calculations of proton affinities as shown by Ozment and
Schmiedekamp
. However, due to computational
expense, application of ab initio methods to the estimation of
proton affinities is still impractical for larger molecules.
Semiempirical methods such as AM1, MNDO and PM3, are not consistently
reliable
in calculations of proton affinities as shown by Ozment and
Schmiedekamp  .
.
The recent progress in the Density Functional Theory (DFT) approaches (for review see refs 4-6) make this method another candidate for reliable calculation of proton affinities, however, the performance of the method in this field is still mostly untested. This prompted us to analyze its performance on a few representative molecules spanning a wide range of proton affinity values.
DFT methods are computationally less demanding than correlated
ab initio approaches and formally scale with the size of the molecule
as  or
or 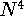 , depending on implementation.
For the the simplest Hartree-Fock ab initio approach the scaling
is . Moreover, the advantage of DFT methods is that they
should in principle include electron correlation energy via
the correlation/exchange potential, while the Hartree-Fock
approach by definition does not include this component of energy.
The simplest of the routinely-used ab initio correlated approaches,
based on the second order many body perturbation theory,
MBPT(2) (frequently called second order Møller-Plesset
theory - MP2), recovers only a portion of the correlation energy and
scales as
, depending on implementation.
For the the simplest Hartree-Fock ab initio approach the scaling
is . Moreover, the advantage of DFT methods is that they
should in principle include electron correlation energy via
the correlation/exchange potential, while the Hartree-Fock
approach by definition does not include this component of energy.
The simplest of the routinely-used ab initio correlated approaches,
based on the second order many body perturbation theory,
MBPT(2) (frequently called second order Møller-Plesset
theory - MP2), recovers only a portion of the correlation energy and
scales as 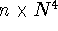 (n is the number of
occupied molecular orbitals).
The major weakness of DFT approaches is that the exact mathematical
form of the exchange-correlation potential is not known. For that reason
approximations are used.
The most popular is the
Local Spin Density (LSD) approximation, which simply assumes that the
exchange-correlation potential dependence upon charge density
is represented by the functional form
found for the homogenous electron gas. This approximation works well in
many cases; however, it suffers from severly underestimating electron
exchange
(n is the number of
occupied molecular orbitals).
The major weakness of DFT approaches is that the exact mathematical
form of the exchange-correlation potential is not known. For that reason
approximations are used.
The most popular is the
Local Spin Density (LSD) approximation, which simply assumes that the
exchange-correlation potential dependence upon charge density
is represented by the functional form
found for the homogenous electron gas. This approximation works well in
many cases; however, it suffers from severly underestimating electron
exchange  and overestimating the correlation energy
and overestimating the correlation energy  . Appropriate
corrections to the local density approximation are therefore sought,
and several of such schemes already exist
. Appropriate
corrections to the local density approximation are therefore sought,
and several of such schemes already exist  .
.