|
pass
|
|
Delta.gif,
Image1.gif,
Image10.gif,
Image11.gif,
Image12.gif,
Image14.gif,
Image15.jpg,
Image2.gif,
Image3.gif,
Image4.gif,
Image5.gif,
Image6.gif,
Image7.gif,
Image8.gif,
Image9.gif,
README,
fig3.jpg,
fig4.jpg,
fig5.jpg,
fig6.jpg,
mailbox.gif,
overview.html,
pass.pdf,
pass10_2.0.36linux.tar.Z,
pass10_2.0.36linux.tar.gz,
pass10_sgi.tar.Z,
pass10_sgi.tar.gz,
pass10_sgi_n32mips3.tar.Z,
pass10_sgi_n32mips3.tar.gz,
pass10_sun.tar.Z,
pass10_sun.tar.gz,
pass_jcamd.html,
pass_usage.html,
passlogo.gif,
redline.gif,
redwhite.gif,
sigma.gif
|
|
|
PASS Usage Syntax
======================================================================
======================================================================
==== PASS - Putative Active Sites with Spheres ======================
============================================= G. Patrick Brady =======
======================================================================
= USAGE: pass ProteinPDBfile =
= =
= where include: =
= =
= <-outdir directory_path> ... Specifies that output (i.e. =
= visualization scripts, PDB files for probes, =
= ASPs, ligands, etc) is to be sent to the =
= specified directory path (defaults to the =
= current directory, ./ ) =
= <-more> ... Identify an enhanced set of probe spheres and =
= active site points (ASPs) =
= <-allprobes> ... Produce a display of all probe spheres =
= (i.e. suppress smoothing which, by default, =
= eliminates any probes that do not have at =
= least smoothcount neighbors lying within =
= smoothradius Angstroms). =
= <-volumes> ... Smooth the final probe spheres, group them =
= via clustering, and compute the volumes of the =
= resulting regions. Outputs several measures =
= of nearness to ligands, in the event that some =
= are present. =
= <-noprobes> ... Do NOT produce a PDB file of all final =
= probe spheres (default is to make this file) =
= <-layers> ... Produce a PDB file of each layer of probe =
= spheres (by default these files are NOT made) =
= <-waters> ... Treat waters as part of the protein (by =
= default, waters are removed and ignored) =
= <-heavyonly> ... Exclude hydrogen atoms from calculation =
= of probes. Default is to sense from input PDB =
= <-hydrogen> ... Include hydrogen atoms in probe calculation =
= <-ligand PDBfilename> ... Read-in a ligand from a =
= separate PDB file (supercedes any HETATM =
= ligand(s) in ProteinPDBfile) =
= Visualization Script Files: load, color, and render the protein, =
= probe spheres, ASPs, and ligand(s). In some cases =
= radial subsets centered on the ASPs are automatically =
= defined. =
= <-cerius2> ... Produce a Cerius2 script (filename.log) =
= <-insightII> ... Produce an InsightII script (filename.bcl) =
= <-moe> ... Produce a MOE script (filename.svl) =
= <-quanta> ... Produce a Quanta script (filename.rec) =
= <-rasmol> ... Produce a RasMol script (filename.ras) =
= <-sybyl> ... Produce a Sybyl script (filename.col) =
======================================================================
|


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
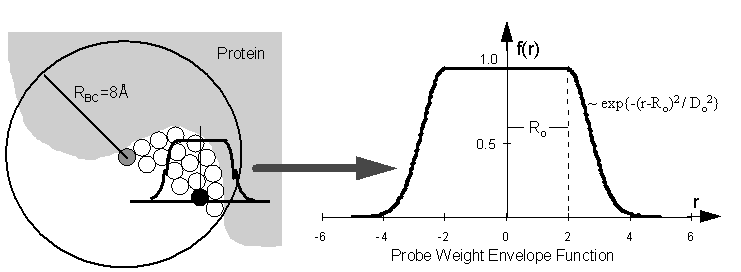{kind=link}
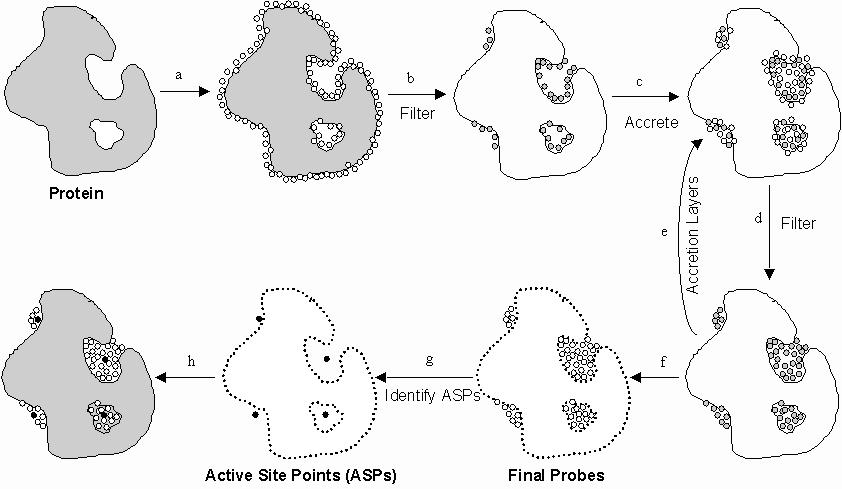{kind=link}
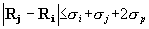{kind=link}
{kind=link}
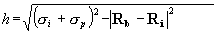{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}