We have shown that the DFT methodology is a good candidate for routine
calculations of proton affinities. However, it is evident that the
nonlocal gradient corrections have to be used to correctly estimate the
change in electronic energy of protonation. The results obtained with
Becke-Perdew corrections applied perturbationally or in a self-consistent
manner are of MP2 quality but require much less computation. Moreover,
since DFT methods scale formally with molecular size as  (compared
to
(compared
to 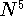 for MP2 ab initio approach) they can be applied to much larger
molecules.
for MP2 ab initio approach) they can be applied to much larger
molecules.
Acknowledgment: Authors are grateful for the Ohio Supercomputer Center grant of computer time which made these calculations possible. JKL, RAH and DDM appreciate the support of National Institutes of General Medical Sciences, GM 29358.