|
dft-overview
|
|
dft-tex,
dft.html,
dft_biol.html,
dft_dft.html,
dft_fof.html,
dft_hamil.html,
dft_hk.html,
dft_intro.html,
dft_perf.html,
dft_refs.html,
dft_scfks.html,
dft_wave.html,
foot_motif.gif,
footnode.html,
image.gif,
img1.gif,
img10.gif,
img100.gif,
img101.gif,
img102.gif,
img103.gif,
img104.gif,
img105.gif,
img106.gif,
img107.gif,
img108.gif,
img109.gif,
img11.gif,
img110.gif,
img111.gif,
img112.gif,
img113.gif,
img114.gif,
img115.gif,
img116.gif,
img117.gif,
img118.gif,
img119.gif,
img12.gif,
img120.gif,
img121.gif,
img122.gif,
img123.gif,
img124.gif,
img125.gif,
img126.gif,
img127.gif,
img128.gif,
img129.gif,
img13.gif,
img130.gif,
img131.gif,
img132.gif,
img133.gif,
img134.gif,
img135.gif,
img136.gif,
img137.gif,
img138.gif,
img139.gif,
img14.gif,
img140.gif,
img141.gif,
img142.gif,
img143.gif,
img144.gif,
img145.gif,
img146.gif,
img147.gif,
img148.gif,
img149.gif,
img15.gif,
img150.gif,
img151.gif,
img152.gif,
img153.gif,
img154.gif,
img155.gif,
img156.gif,
img157.gif,
img158.gif,
img159.gif,
img16.gif,
img160.gif,
img161.gif,
img162.gif,
img163.gif,
img164.gif,
img165.gif,
img166.gif,
img167.gif,
img168.gif,
img169.gif,
img17.gif,
img170.gif,
img171.gif,
img172.gif,
img173.gif,
img174.gif,
img175.gif,
img176.gif,
img177.gif,
img178.gif,
img179.gif,
img18.gif,
img180.gif,
img181.gif,
img182.gif,
img183.gif,
img184.gif,
img185.gif,
img186.gif,
img187.gif,
img188.gif,
img189.gif,
img19.gif,
img190.gif,
img191.gif,
img192.gif,
img193.gif,
img194.gif,
img195.gif,
img196.gif,
img197.gif,
img198.gif,
img199.gif,
img2.gif,
img20.gif,
img200.gif,
img201.gif,
img202.gif,
img203.gif,
img204.gif,
img205.gif,
img206.gif,
img207.gif,
img208.gif,
img209.gif,
img21.gif,
img210.gif,
img211.gif,
img212.gif,
img213.gif,
img214.gif,
img215.gif,
img216.gif,
img217.gif,
img218.gif,
img219.gif,
img22.gif,
img220.gif,
img221.gif,
img222.gif,
img223.gif,
img224.gif,
img225.gif,
img226.gif,
img227.gif,
img228.gif,
img229.gif,
img23.gif,
img230.gif,
img231.gif,
img232.gif,
img233.gif,
img234.gif,
img235.gif,
img236.gif,
img237.gif,
img238.gif,
img239.gif,
img24.gif,
img240.gif,
img241.gif,
img242.gif,
img243.gif,
img244.gif,
img245.gif,
img246.gif,
img247.gif,
img248.gif,
img249.gif,
img25.gif,
img250.gif,
img251.gif,
img252.gif,
img253.gif,
img254.gif,
img255.gif,
img256.gif,
img26.gif,
img27.gif,
img28.gif,
img29.gif,
img3.gif,
img30.gif,
img31.gif,
img32.gif,
img33.gif,
img34.gif,
img35.gif,
img36.gif,
img37.gif,
img38.gif,
img39.gif,
img4.gif,
img40.gif,
img41.gif,
img42.gif,
img43.gif,
img44.gif,
img45.gif,
img46.gif,
img47.gif,
img48.gif,
img49.gif,
img5.gif,
img50.gif,
img51.gif,
img52.gif,
img53.gif,
img54.gif,
img55.gif,
img56.gif,
img57.gif,
img58.gif,
img59.gif,
img6.gif,
img60.gif,
img61.gif,
img62.gif,
img63.gif,
img64.gif,
img65.gif,
img66.gif,
img67.gif,
img68.gif,
img69.gif,
img7.gif,
img70.gif,
img71.gif,
img72.gif,
img73.gif,
img74.gif,
img75.gif,
img76.gif,
img77.gif,
img78.gif,
img79.gif,
img8.gif,
img80.gif,
img81.gif,
img82.gif,
img83.gif,
img84.gif,
img85.gif,
img86.gif,
img87.gif,
img88.gif,
img89.gif,
img9.gif,
img90.gif,
img91.gif,
img92.gif,
img93.gif,
img94.gif,
img95.gif,
img96.gif,
img97.gif,
img98.gif,
img99.gif
|
|
|
Molecular DFT
Introduction to Molecular Approaches of Density Functional Theory
Ohio Supercomputer Center, 1224 Kinnear Rd, Columbus, OH 43221-1153
Return to index
Advantages of DFT in biological chemistry
- Computational demands are much less severe than
for ab initio methods of similar quality - hence
the method is applicable to much larger molecules.
- Metals are frequently present in active centers
of enzymes. Traditional ab initio methods have
severe problems with transition metals. In fact, it can
be proved that Hartree-Fock equation cannot be solved for
the true metalic state. It is related to the fact that
there is a difficulty to converge HF when highest occupied
orbitals are very close in energy (the situation very
popular for transition metals).
- The DFT, similar to ab initio methods,
is nonparametric, i.e., applicable to any molecule.
While some say that basis sets are parameters
for ab initio and DFT methods, this is an
exaggeration. Basis sets are easily derived from
atomic calculations, and beside, they were
derived long time ago for all elements of periodic
table.
- The restriction of DFT being applicable to
the ground state only is not usually a problem, unless
you study interaction of radiation with biological
molecules (e.g., UV induced mutations).
Return to index
REFERENCES
|


{kind=link}
{kind=link}
{kind=link}
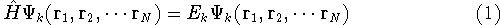{kind=link}
{kind=link}
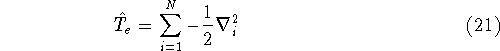{kind=link}
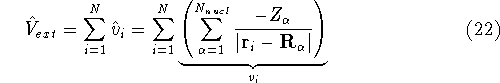{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
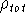{kind=link}
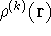{kind=link}
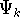{kind=link}
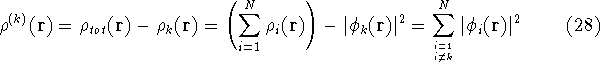{kind=link}
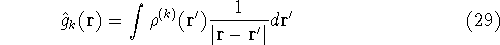{kind=link}
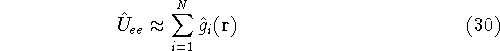{kind=link}
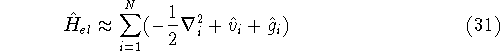{kind=link}
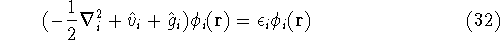{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
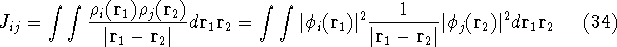{kind=link}
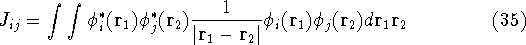{kind=link}
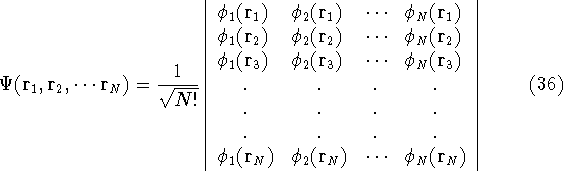{kind=link}
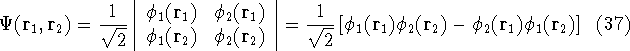{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
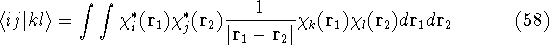{kind=link}
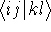{kind=link}
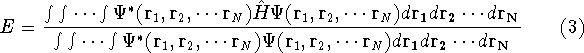{kind=link}
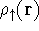{kind=link}
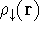{kind=link}
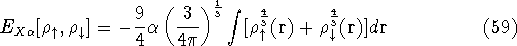{kind=link}
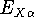{kind=link}
{kind=link}
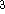{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
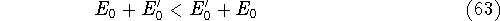{kind=link}
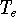{kind=link}
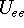{kind=link}
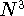{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
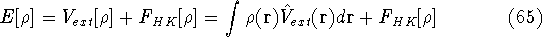{kind=link}
{kind=link}
{kind=link}
{kind=link}
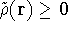{kind=link}
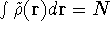{kind=link}
{kind=link}
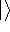{kind=link}
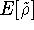{kind=link}
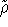{kind=link}
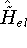{kind=link}
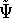{kind=link}
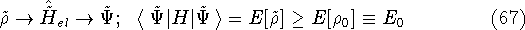{kind=link}
{kind=link}
{kind=link}
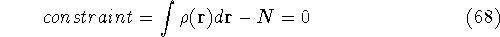{kind=link}
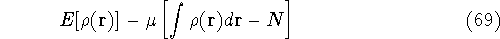{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
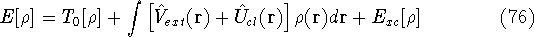{kind=link}
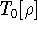{kind=link}
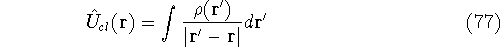{kind=link}
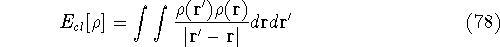{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
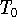{kind=link}
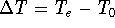{kind=link}
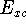{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
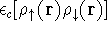{kind=link}
{kind=link}
{kind=link}
{kind=link}
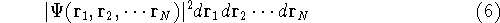{kind=link}
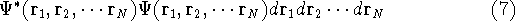{kind=link}
{kind=link}
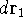{kind=link}
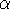{kind=link}
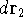{kind=link}
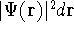{kind=link}
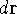{kind=link}
{kind=link}
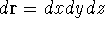{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
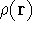{kind=link}
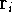{kind=link}
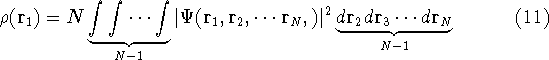{kind=link}
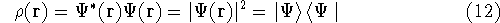{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
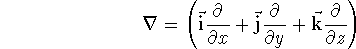{kind=link}
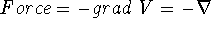{kind=link}
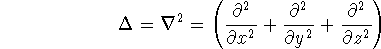{kind=link}
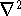{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
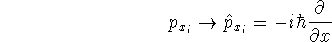{kind=link}
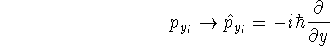{kind=link}
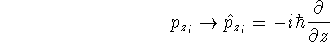{kind=link}
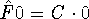{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
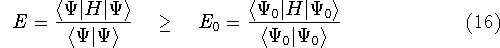{kind=link}
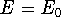{kind=link}
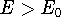{kind=link}
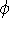{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
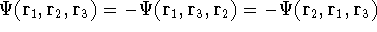{kind=link}
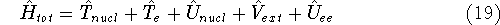{kind=link}
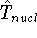{kind=link}
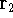{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}