|
dft-overview
|
|
dft-tex,
dft.html,
dft_biol.html,
dft_dft.html,
dft_fof.html,
dft_hamil.html,
dft_hk.html,
dft_intro.html,
dft_perf.html,
dft_refs.html,
dft_scfks.html,
dft_wave.html,
foot_motif.gif,
footnode.html,
image.gif,
img1.gif,
img10.gif,
img100.gif,
img101.gif,
img102.gif,
img103.gif,
img104.gif,
img105.gif,
img106.gif,
img107.gif,
img108.gif,
img109.gif,
img11.gif,
img110.gif,
img111.gif,
img112.gif,
img113.gif,
img114.gif,
img115.gif,
img116.gif,
img117.gif,
img118.gif,
img119.gif,
img12.gif,
img120.gif,
img121.gif,
img122.gif,
img123.gif,
img124.gif,
img125.gif,
img126.gif,
img127.gif,
img128.gif,
img129.gif,
img13.gif,
img130.gif,
img131.gif,
img132.gif,
img133.gif,
img134.gif,
img135.gif,
img136.gif,
img137.gif,
img138.gif,
img139.gif,
img14.gif,
img140.gif,
img141.gif,
img142.gif,
img143.gif,
img144.gif,
img145.gif,
img146.gif,
img147.gif,
img148.gif,
img149.gif,
img15.gif,
img150.gif,
img151.gif,
img152.gif,
img153.gif,
img154.gif,
img155.gif,
img156.gif,
img157.gif,
img158.gif,
img159.gif,
img16.gif,
img160.gif,
img161.gif,
img162.gif,
img163.gif,
img164.gif,
img165.gif,
img166.gif,
img167.gif,
img168.gif,
img169.gif,
img17.gif,
img170.gif,
img171.gif,
img172.gif,
img173.gif,
img174.gif,
img175.gif,
img176.gif,
img177.gif,
img178.gif,
img179.gif,
img18.gif,
img180.gif,
img181.gif,
img182.gif,
img183.gif,
img184.gif,
img185.gif,
img186.gif,
img187.gif,
img188.gif,
img189.gif,
img19.gif,
img190.gif,
img191.gif,
img192.gif,
img193.gif,
img194.gif,
img195.gif,
img196.gif,
img197.gif,
img198.gif,
img199.gif,
img2.gif,
img20.gif,
img200.gif,
img201.gif,
img202.gif,
img203.gif,
img204.gif,
img205.gif,
img206.gif,
img207.gif,
img208.gif,
img209.gif,
img21.gif,
img210.gif,
img211.gif,
img212.gif,
img213.gif,
img214.gif,
img215.gif,
img216.gif,
img217.gif,
img218.gif,
img219.gif,
img22.gif,
img220.gif,
img221.gif,
img222.gif,
img223.gif,
img224.gif,
img225.gif,
img226.gif,
img227.gif,
img228.gif,
img229.gif,
img23.gif,
img230.gif,
img231.gif,
img232.gif,
img233.gif,
img234.gif,
img235.gif,
img236.gif,
img237.gif,
img238.gif,
img239.gif,
img24.gif,
img240.gif,
img241.gif,
img242.gif,
img243.gif,
img244.gif,
img245.gif,
img246.gif,
img247.gif,
img248.gif,
img249.gif,
img25.gif,
img250.gif,
img251.gif,
img252.gif,
img253.gif,
img254.gif,
img255.gif,
img256.gif,
img26.gif,
img27.gif,
img28.gif,
img29.gif,
img3.gif,
img30.gif,
img31.gif,
img32.gif,
img33.gif,
img34.gif,
img35.gif,
img36.gif,
img37.gif,
img38.gif,
img39.gif,
img4.gif,
img40.gif,
img41.gif,
img42.gif,
img43.gif,
img44.gif,
img45.gif,
img46.gif,
img47.gif,
img48.gif,
img49.gif,
img5.gif,
img50.gif,
img51.gif,
img52.gif,
img53.gif,
img54.gif,
img55.gif,
img56.gif,
img57.gif,
img58.gif,
img59.gif,
img6.gif,
img60.gif,
img61.gif,
img62.gif,
img63.gif,
img64.gif,
img65.gif,
img66.gif,
img67.gif,
img68.gif,
img69.gif,
img7.gif,
img70.gif,
img71.gif,
img72.gif,
img73.gif,
img74.gif,
img75.gif,
img76.gif,
img77.gif,
img78.gif,
img79.gif,
img8.gif,
img80.gif,
img81.gif,
img82.gif,
img83.gif,
img84.gif,
img85.gif,
img86.gif,
img87.gif,
img88.gif,
img89.gif,
img9.gif,
img90.gif,
img91.gif,
img92.gif,
img93.gif,
img94.gif,
img95.gif,
img96.gif,
img97.gif,
img98.gif,
img99.gif
|
|
|
Molecular DFT
Introduction to Molecular Approaches of Density Functional Theory
Ohio Supercomputer Center, 1224 Kinnear Rd, Columbus, OH 43221-1153
Return to index
INTRODUCTION
There are many approaches of computational chemistry which are popular
in molecular modeling:
- Simple Comparative and Graphical Approaches - graphical inspection,
molecular superposition, overlapping/nonoverlapping volume, topological
indices, traditional SAR and QSAR, rigid conformational search, ComFA, shape
analysis, etc. Used as a first step in scanning biologically active
molecules and useful in detecting characteristic molecular
features needed for activity. These methods are not quantitative,
since they do not consider energetics of receptor-ligand
interactions.
- Empirical approaches - molecular mechanics,
molecular dynamics. Relatively simple interatomic potentials,
electrostatic interactions, and dispersion forces, allow for
basic comparisons of energetics and geometry optimization.
Solvent effects can be included explicitly or via empirical models.
Very useful and fast compared to rigorous quantum calculations.
Major drawbacks: experimental or theoretically derived information
is needed to ``standardize''
models and parameters. In principle, these approaches are not able to
model chemical reactions, bond forming/breaking since electronic structure
does not enter these models.
- Quantum Approaches - based on explicit consideration
of the electronic structure. These methods are substantially
more computationally demanding then comparative and empirical
approaches for the molecules of the same size. They can be roughly
divided into:
- Semiempirical methods - approximate methods in which
some quantities are taken from experiment, some small quantities
are neglected, and some quantities estimated by fitting to experimental
data. May only be used for chemical species for which they were parameterized.
For distorted, uncommon bonding situations produce unreliable results.
- Nonempirical methods - do not require empirical parameters
and can be used for any molecular system.
- Traditional ab initio - use Hartree-Fock method as a starting
point, i.e., wave function is used to describe electronic structure.
- Density Functional Methods - electron density as a
primary way of describing the system.
There is a justified interest in Density Functional Approaches among chemists.
While they have been a domain of physicists for a long time, these methods have now
found their way into mainstream chemistry. The traditional ab initio
approaches, which are the workhorse of Quantum Chemistry offer a prescription
for calculated truth. In principle, one can calculate chemical properties
with any desired accuracy. The methods are known and proven to work.
The only problem is that for systems larger than hydrogen or lighter HnX molecules,
the calculations involved in obtaining accurate results are frequently impractical.
We know how to compute it, but we do not have computational power to do it (probably
for many years to come, even with the spectacular progress in computer technology).
For this reason, the contemporary research in traditional ab initio methods is
concerned mainly with better approximations to a Full CI method, or infinite order
perturbational expansions, which would give good quality reliable results
with reasonable computational effort. Still, the most promising Coupled Cluster (CC)
and Complete Active Space SCF (CASSCF) calculations scale more than 5th power
in molecular size, and are impractical for molecules containing tens of atoms.
Density Functional Theory does not provide a prescription how to calculate
truth. It only provides the existence proof for possibility of obtaining
accurate results, but no prescription for systematic improvement. The DFT
is accurate, if we knew how to derive necessary relations between density
and energy. Unfortunately,
energy functionals relating electronic density to energy are unknown,
and there is no general way to improve them beside trying new ones and
judging their quality by the results. As Prof. Perdew once said:
"We are like a blind person who wants to find out how the elephant looks like
by touching its legs" (sorry, the quotation is from memory, and may not
be accurate). But the Density Functional Theory provides a hope for a method
which scales with the size as 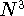 in the worst case, and possibly linearly for
larger molecules (Zhou, 1995; Yang, 1991). So it is used in the
spirit of late prof. Slater saying (about his X in the worst case, and possibly linearly for
larger molecules (Zhou, 1995; Yang, 1991). So it is used in the
spirit of late prof. Slater saying (about his X 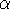 method): Do you want to
calculate it, or do you want it to be accurate? The DFT results are in many
cases surprisingly good if one takes into account the crude approximations
on which some of them are based. For example, Local Spin Density calculations
yield results on many molecular properties which are of quality comparable
with higher order ab initio correlated methods, yet, the LSD assumes that
electrons in molecules behave like electrons in an uniform electron gas.
Moreover, DFT methods frequently provide reliable answer for cases which are
especially difficult to address with conventional ab initio methods,
like, e.g., transition metals. On the other hand, they frequently fail miserably,
e.g., in charge-transfer complexes. method): Do you want to
calculate it, or do you want it to be accurate? The DFT results are in many
cases surprisingly good if one takes into account the crude approximations
on which some of them are based. For example, Local Spin Density calculations
yield results on many molecular properties which are of quality comparable
with higher order ab initio correlated methods, yet, the LSD assumes that
electrons in molecules behave like electrons in an uniform electron gas.
Moreover, DFT methods frequently provide reliable answer for cases which are
especially difficult to address with conventional ab initio methods,
like, e.g., transition metals. On the other hand, they frequently fail miserably,
e.g., in charge-transfer complexes.
The fact that more, and more ab initio packages provide options for
DFT calculations, is a sign of changing times, and the indication that
even the most vigorous opponents of this method see that it is just
another way of doing things. On the other hand, DFT is in principle only
applicable to the ground state, and there is little hope that it will
be extended in a practical way to excited states in a straightforward manner
any time soon.
Return to index
Next Section
|


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
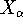{kind=link}
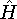{kind=link}
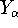{kind=link}
{kind=link}
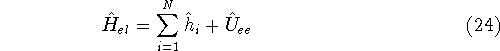{kind=link}
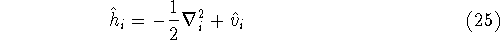{kind=link}
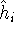{kind=link}
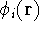{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
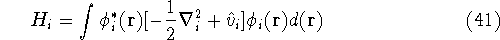{kind=link}
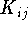{kind=link}
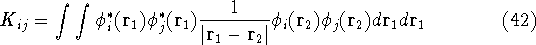{kind=link}
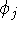{kind=link}
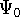{kind=link}
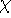{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
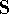{kind=link}
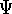{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
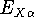{kind=link}
{kind=link}
{kind=link}
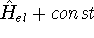{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
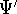{kind=link}
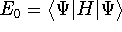{kind=link}
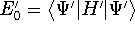{kind=link}
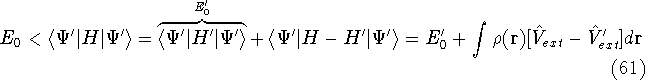{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
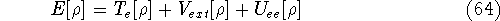{kind=link}
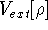{kind=link}
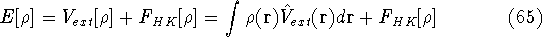{kind=link}
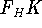{kind=link}
{kind=link}
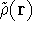{kind=link}
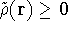{kind=link}
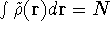{kind=link}
{kind=link}
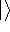{kind=link}
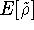{kind=link}
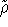{kind=link}
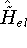{kind=link}
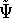{kind=link}
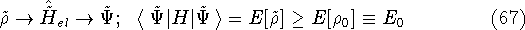{kind=link}
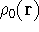{kind=link}
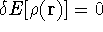{kind=link}
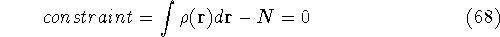{kind=link}
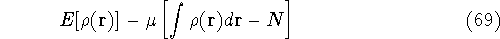{kind=link}
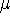{kind=link}
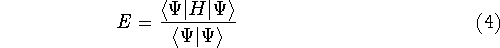{kind=link}
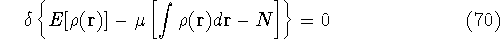{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
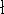{kind=link}
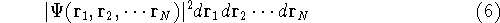{kind=link}
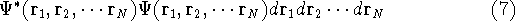{kind=link}
{kind=link}
{kind=link}
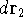{kind=link}
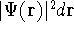{kind=link}
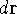{kind=link}
{kind=link}
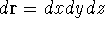{kind=link}
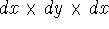{kind=link}
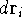{kind=link}
{kind=link}
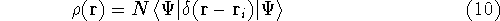{kind=link}
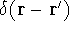{kind=link}
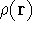{kind=link}
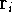{kind=link}
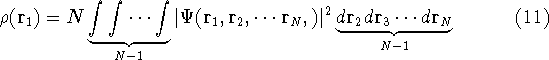{kind=link}
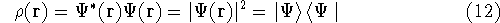{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
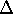{kind=link}
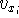{kind=link}
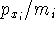{kind=link}
{kind=link}
{kind=link}
{kind=link}
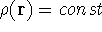{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
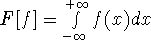{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
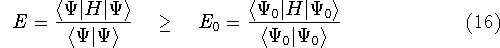{kind=link}
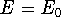{kind=link}
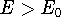{kind=link}
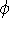{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
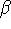{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}